
Ever since my time in a crystallography lab, I’ve been fascinated by the process of translating raw diffraction patterns into detailed structural models.

Back then, we relied on those cool SGI (Silicon Graphics, Inc.) computers—high-performance machines that were the go-to for graphics and visualization in the late ’90s and early 2000s. These workstations were the backbone of many scientific labs, enabling us to visualize complex molecular structures in ways that were groundbreaking at the time.
Even at that time, I was captivated by the pursuit of renders that were more than just scientific models—they were almost eye candy, visually arresting representations of the intricate dance of atoms. These weren’t just static arrangements; they were the keys to understanding quantum-scale processes. I found myself wondering: What do these molecular machines really look like? Are they transparent? Gooey?
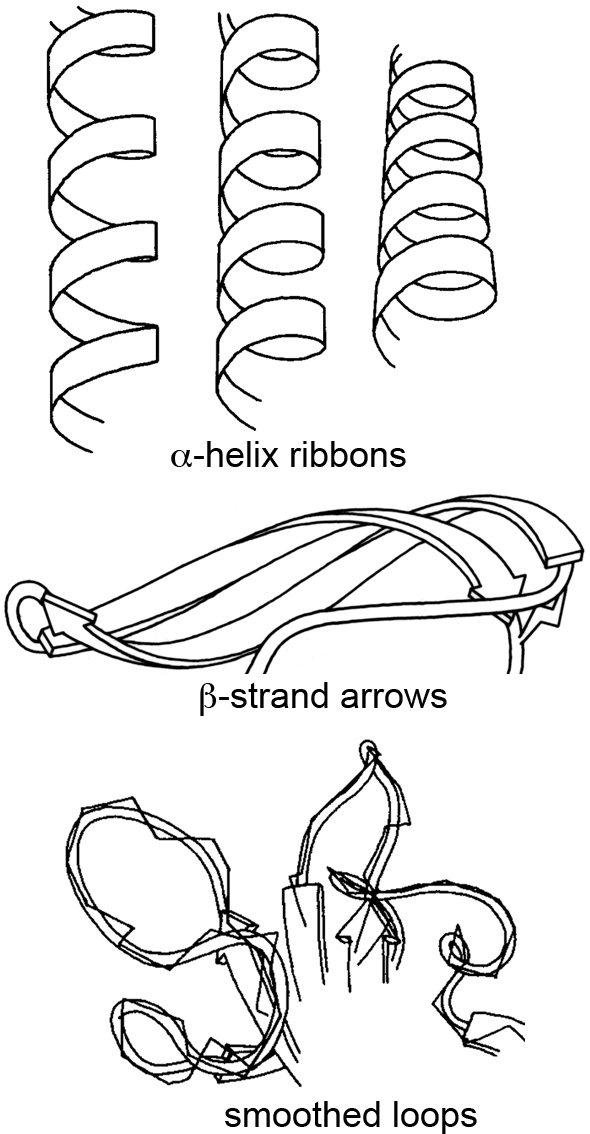
At the time, most people stuck with the ball-and-stick method of molecular rendering, and sometimes the very creative ribbon method for visualizing secondary structures like α-helices and β-sheets. This ribbon method, first introduced by Jane Richardson in the 1980s, revolutionized how we perceive and depict the elegant architecture of proteins.
I was particularly struck by a talk she gave a few years ago at the Machine Learning in Structural Biology (MLSB) workshop, which is part of the broader NeurIPS (Conference on Neural Information Processing Systems). The presentations at MLSB highlight how the intersection of machine learning and structural biology is opening up new avenues for visualizing and understanding complex biological data—building on the foundations researchers laid with creative solutions like the ribbon diagrams decades earlier. You can still view a recording of the entire Richardson talk here.

Fast forward a few years, and I find myself reminiscing about the times when Sonic Youth was the soundtrack to my late-night coding sessions, and I was drawn to ideas about being in the flow, letting go, and just being present in the moment. These days, hanging out with my wife and child brings that same sense of peace and connection. But as much as I cherish these moments, there’s something uniquely exhilarating about attending conferences, where you get to “talk shop” and be exposed to the latest innovations.
I remember the first time I saw molecular renderings by Drew Berry—it was at a VIZBI conference, a gathering that perfectly blends scientific rigor with creative visualization. VIZBI (Visualizing Biological Data) is more than just a conference; it’s an international meeting point for the best minds in science, bioinformatics, and data visualization. What makes VIZBI special is its emphasis on both the scientific and artistic aspects of data visualization.

The conference not only showcases cutting-edge visualizations that transform how life scientists view and interact with data, but it also encourages a deeper appreciation for the aesthetic quality of these visualizations. This was evident in Drew Berry’s work, which brought the molecular world to life in a way I had never seen before. The structures didn’t just sit there; they vibrated, darted around, and had this incredible “stochastic” feel to them, capturing the chaotic energy that defines molecular interactions. It was like seeing the molecules not just as static models, but as living entities, each with its own rhythm and motion. VIZBI isn’t just about keeping up with the latest research; it’s about being inspired, about seeing the boundaries of science and art blur in ways that open up new possibilities for how we understand life at its most fundamental level. It’s the kind of experience that reminds me why I got into this field in the first place.
In the last couple of years, BioVis has really stepped up its efforts to engage the community, working closely with other organizations like IEEE and ISCB to keep the spirit vibrant and interdisciplinary. BioVis has taken on the challenge of pushing forward the frontiers of biological data visualization, encouraging collaboration across fields and nurturing a community that is as diverse as it is innovative.

By bringing together visualization researchers with biologists and bioinformaticians, BioVis has managed to keep the conversation fresh and evolving, ensuring that new methods and ideas keep flowing. It’s exciting to see how these gatherings—both old and new—continue to energize the community and drive progress in understanding the complexities of life at every scale.
And hey, these are just some of the meetings out there, but they are my personal favorites. Even with incredible advancements like AlphaFold, RoseTTAFold, and ColabFold, which have made huge leaps in predicting molecular structures, there is still something uniquely thrilling about the art of representation. For me, that thrill is often fueled by the same sense of awe I get from playing video games. A good game isn’t just about throwing the newest engine at it; it’s about the aesthetics, the art, and the way it all comes together. That’s what makes good games age well—and I think it’s the same with science.



As the saying goes: sometimes, it’s less about where the path leads and more about the wonder found along the way.


















