Over christmas the Genome Reference Consortium gave all of us doing in silico life-science a wonderful present, or maybe it was just a lump of coal. GRCh38, the newest human reference genome assembly, was released to cheers and jeers abound.
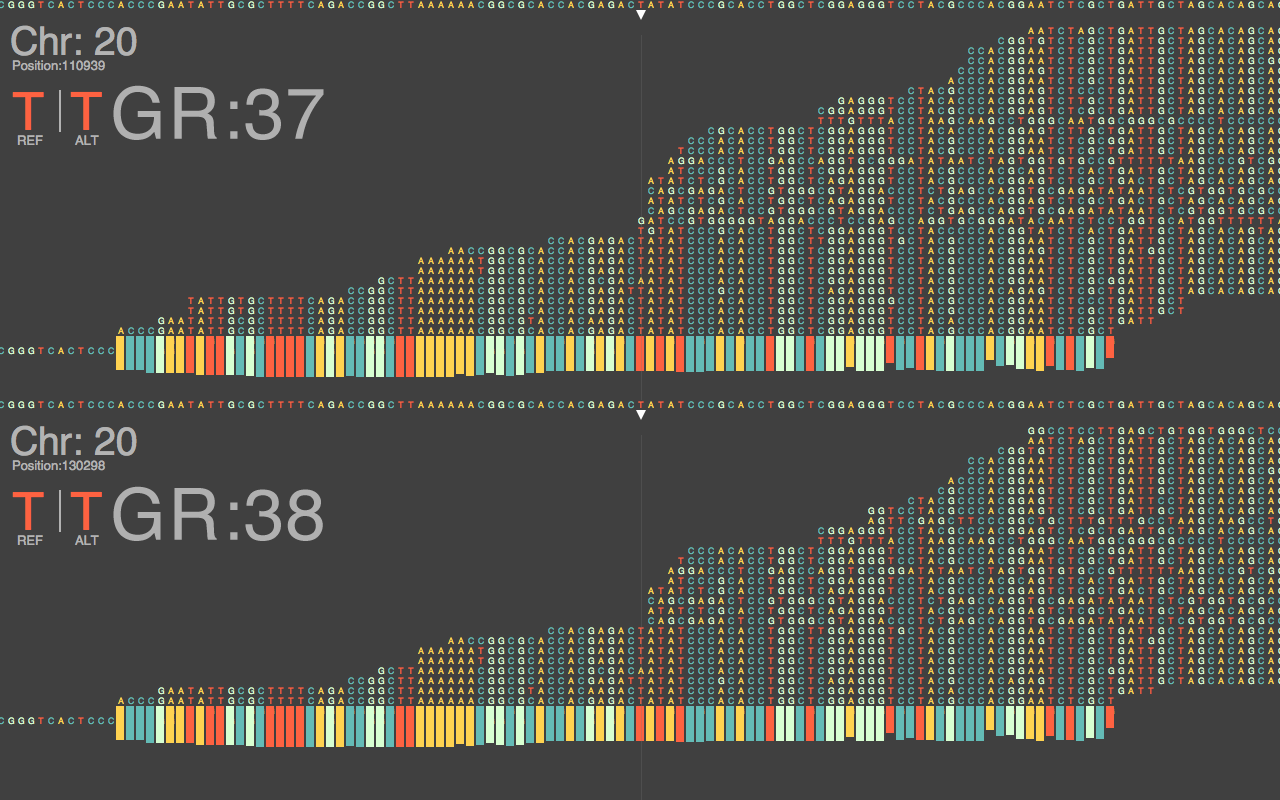
Fig.1: Chromosome 20 Assembly With BWA
Of course, most of us are excited to have up-to-date standards, especially with something like the human reference playing such a pivotal role in clinical adoption of genomics. However, some are lamenting the perhaps inevitable remapping of their NGS reads to this new reference. And it’s completely reasonable to have this worry. Will previous results from samples remain valid once assembled with this new reference, and how different will the sequence alignments be?
Fig.2: Ts/Tv Ratios between GRCh37 & 38
It will take months and years to thoroughly answer these questions, and notice the complete impact of this update on current NGS data. Consequentially, this does not keep us from starting to get preliminary ideas of what to expect in the coming years of working with this build. Figure 1 above, shows a nucelotide-level closeup of a BWA sequence alignment of the same dataset, generated on a HiSeq 2500, across the last major release of GRCh37 and the new GRCh38.
Internally we have been using chromosome 20 of human reference builds to benchmark tools and pipelines with datasets; it has a favorable size in terms of length, and not too many curveballs in terms of features. Figure 2 to the left, shows the Ts/Tv ratios between the two alignments of the same data across the two references to be quite similar at 0.3527 and 0.3445, respectively. Working with the slew of aligners, BWA has repeatedly shown itself to produce reliable results, while avoiding any overly-complex algorithms and trendy implementations. It’s a good workhorse.
Similarly, samtools was used to parse through our SAM/BAM files to produce VCFs with mpileup. Which, again, does not have the most bells and whistles, but is consistently reliable and good for comparing a single variable, in this case, the reference.
Fig.3: High-level Alignment Map Topology
Quantifiably, GRCh38 is very similar to the later GRCh37 releases, showing a change rate of 1 change every 159,558 bases on 37.69 and 1 change every 156,779 bases on 38 for our chromosome 20 dataset. Which, to use a technical term, looks pretty damn close. One of the major updates according to the GRC are changes to chromosome coordinates, which some back of the envelope math seems to give a Δ of +19,359 between GRCh37.69 and 38. In combination with one of the other major updates, sequence representation for centromeres, short-reads appear to be spread thinner to cover this difference, resulting in slightly lower depth of coverage versus 37.69. Figure 3 above, shows that overall the alignment map remains mostly similar, at least when BWA is used with standard Illumina reads; with somewhat negligible loss of DP.
Fig.4: Chr20 Annotation of Regional Features
By far the largest noticeable change brought to preexisting datasets appears to be related to annotation. Figure 4 above, shows just how hopelessly incongruent GRCh38 is at the moment with current annotation resources, yielding the largest differences to the latest GRCh37 assembly for the same reads. But this was to be expected, annotation will very likely be the last to catch up to this new build, and will improve over months and years.
So, should your project start using GRCh38? The short answer is, not yet. The long answer, it depends on your project resources, pipeline flexibility, and the questions trying to be answered. It’s helpful to remap preexisting NGS data to this new reference, and newly generated datasets would benefit the most, as sequence alignment tends to be the most expensive process in the pipeline to redo. Just keep in mind that for useful results your entire analysis pipeline, which is often an amalgamation of various opensource, commercial, and internal components has to work together. For the time being, GRCh38 is a wrench in the gears for many people, but it has a very promising future.
While the UnifiedGenotyper included within the Genome Analysis Toolkit (GATK) provides an ample method by which to call SNPs and indels, mpileup within Samtools still remains a reliable, quick and straightforward way to get variants.
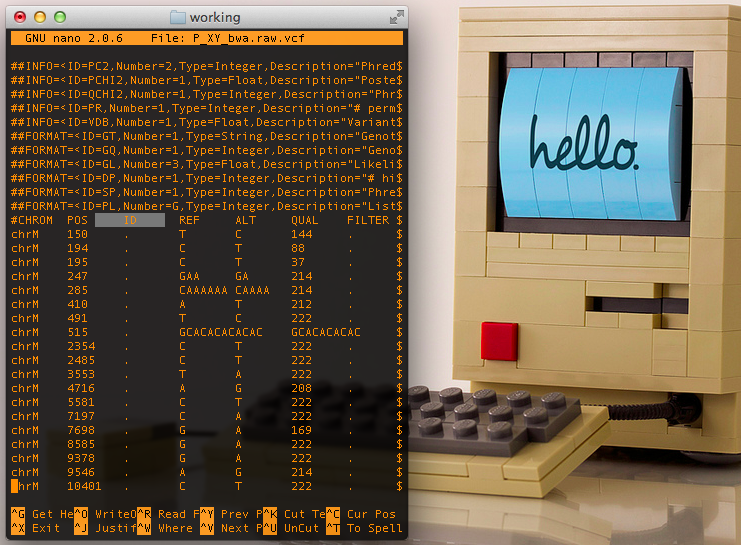
Raw VCF file from Samtools, notice lack of annotations & filters in the 3rd and 7th columns
To begin we take our assembled bam files created by the method of your choice, two of which are described in the previous posts[1][2]. With newer versions of Samtools the pileup function is replaced by mpileup, they perform the exact same actions; however, in traditional pileup we pass a single individual genome as a bam file for variant discovery, while in mipleup we can pass multiple individuals together and each of their variants are discovered within a single file as the output.
Even though we’re saying the variant discovery is by samtools, all the actual work is being done by bcftools. To learn more about what bcftools can do check out the documentation, all the modules are included as a subdirectory within the samtools package.
Now that we have a VCF file containing all the positions where our samples differ from the reference, and each other, we can begin to utilize the appropriate GATK modules. Starting with annotation:
As you can see these one liners can get quite long, but rest assured, the results are worthwhile. If you look carefully at the above command you can see that we’re annotating based on a second VCF file, which in this case is being attained from the NCBI’s dbSNP. Feel free to use whatever database you see fit to generate your annotations.
Annotated VCF, notice rsIDs in 3rd column
Annotating our raw VCF with a dbSNP file results in flagging any polymorphisms between our sets to be marked with an rsID. These unique identifiers are used to track individual disease phenotypes, which are at various points of experimental validation. However, if we take our mapped genome and search for variations we’ll soon find that there are simply too many variations that show up to make any sense of our data. We have to decrease the size of our haystack before we start looking for our needle. This is where Filtration comes in. A high-level overview of the process can be seen in this previous post which utilizes a key figure from Genomics & Computation (available on iTunes). Below we execute a part of these concepts using GATK:
It is important to understand the one-to-one mapping of filtering expression to the filter name to adequately use this module. A filterExpression should take any number of fields available within the INFO field for any given variation, such as:
For example the expression could take into account the depth of read as well as the mapping quality, stating & . However these expressions have to be written in Java Expression Language (JEXL) and are then mapped directly to the following filterName, multiple expression/name combinations can be linked in a single pipe.
There are many more steps towards refinement, i.e. recalibration and variant selection, but this blog post is getting quite long. And I think if you follow the roadsigns laid out here the full abilities of both Samtools & the GATK will become evident. The final payoff being reliable, meaningful, and thus useful, NGS data. Hit me up if you get stuck or think my ways are lame.
Genomic code that makes us is made up of four letters, ATGC. Billions of these letters together creates a lifeform. Iterated function systems (IFS) are anything that can be made by repeating the same simple rules over and over. The easiest example being tree branches, add a simple structure repeatedly ad-infinitum and before you know it we have complex and beautiful systems; the popular example being the Sierpinski Triangle or “triforce” for the Zelda fans. As the cost of DNA sequencing becomes cheaper day by day we are confronted with a tsunami of data and it has become exceedingly difficult to derive meaningful answers from all the information contained within us.
H. Sapiens
Finding any advantage in ways to organize and view the data helps us discover minuet differences between individuals or say a normal cell versus a cancer cell. This is where Chaos Game Representation (CGR) becomes helpful, CGR is just a form of IFS that is helpful in mapping seemingly random information, that we suspect or know to have some sort of underlying structure.
In our case this would be the human genome. Although when looking at the letters coming from our DNA it seems like billions of random babbles, it is of course organized in a manner to give the blueprint for our bodies. So let’s roll the dice- do we get any sort of meaningful structure when applying CGR to DNA? If you are so inclined, something fun to try is the following:
For example, reading the sequence in order, apply T1 whenever C is encountered, apply T2 whenever A is encountered, apply T3 whenever T is encountered, and apply T4 whenever G is encountered. Really though any transformations to C, A, T, and G can be used and multiple methods can be compared. Self-similarity is immediately noticeable in these maps, which isn’t all that surprising since fractalsare abundant in nature and DNA after all, is a natural syntax. Being aware that these patterns exist within our data, opens us up to some new questions to evaluate if IFS, CGR and fractals in general are helpful tools in the interpretation of genomic data.
Signal transducer 5B (STAT5B), on chromosome 17
Since the mapping is 1-1 and we see patterns emerge, we are hinted that there may be biological relevance; especially because different genes yield different patterns. But what exactly are the correlations between the patterns and the biological functions? It would also be very interesting to see mappings of introns/exons colored differently or color amino acids and various codons. One thing is for sure, genomes aren’t just endless columns and rows of letters, they are pictures. It is much easier to compare pictures and discover variations, which can ultimately allow us to find meaningful interpretation from this invaluable data.
Citations:
Jeffrey, H. J., “Chaos game visualization of sequences,” Computers & Graphics 16 (1992), 25-33.
Ashlock, D. Golden, J.B., III. Iterated function system fractals for the detection and display of DNA reading frame (2000) ISBN: 0-7803-6375-2
VV Nair, K Vijayan, DP Gopinath ANN based Genome Classifier using Frequency Chaos Game Representation (2010)
Version 0.1 the first print from github.com/otaviogood
I have a kid now, and so do a few people I’ve worked closely with for years. We started talking about sampling bias—not in models, but in life. The neighborhoods we’re raising our kids in, places like Palo Alto and the Santa Monica Mountains, are extreme outliers. The people our children interact with every day are a narrow slice of the global population. That shared realization eventually turned into a question: what would it look like to generate a more honest sample of humanity—and what if you turned it into a game?
🎲 Using deterministic random seed:1979
🌟 Starting Program 1:Position Sampling and Data Fetching...
📡 Fetching Wage and salaried workers%(attempt 1/3):https://api.worldbank.org/v2/country/DE/indicator/SL.EMP.WORK.ZS?format=json&per...
✅ Position sampling and data fetching complete!
✅ Successfully fetched Gini coefficient
The idea first surfaced when a friend and longtime collaborator told me he’d been trying to make a game for his kids. The motivation was there, but the early results were unsettling. The sampling was honest, but not gentle: one of the first characters it produced was a visibly sad child in war-torn Ukraine. Others leaned uncomfortably into stereotypes. He builds games for kids all the time, and this felt different—not wrong, exactly, but revealing. The system wasn’t misbehaving; it was reflecting something real.
To deal with that, we started talking about history—not as a backdrop, but as a variable. The uncomfortable cards were snapshots, frozen at a single moment in time. But human societies aren’t static. Places that look prosperous today were often miserable in the past, and places struggling now have gone through long periods of stability, creativity, or power. Bringing in historical context let us shift from a flat sample of “now” to something longitudinal. It also opened a way to teach kids that human and cultural development isn’t a snapshot—it’s a time series.
Wikipedia felt like a good place to start. I’ve been reading history there for years, and it’s one of the few places where human knowledge is both deep and structured enough to be programmatically useful. The system starts by crawling Wikipedia’s historical eras portal and generating JSON files of eras across human history. When the sampler picks a location, it finds which eras align and uses those as structured seeds. By recursively walking category graphs and pulling summaries, the system generates people who make sense in their time: era-appropriate names, occupations, interests, stories, and images. Each era becomes a bounded distribution rather than a vibe, constraining technologies, social roles, and narratives. The LLM isn’t inventing history—it interpolates within it. History stops being static and becomes a generator, turning the game into a longitudinal model of humanity instead of a snapshot of the present.
// Recursively fetch articles from a category and its subcategories
description:"The American Wild West during the late 19th century, known for cowboys, frontier towns, gold rushes, and interactions between settlers and Native Americans.",
description:"The Vandals were a Germanic people living in the forests and plains of Germania during the late Roman Empire. Known for their fierce warrior culture, tribal councils, and migrations, they clashed with Roman legions and other tribes. Life was marked by hunting, raiding, feasting, and storytelling around the fire. Villages were ruled by chieftains, and society was patriarchal, with women managing homes and resources. The Vandals practiced pagan rituals, revered their ancestors, and relied on skilled blacksmiths and horsemen. Their reputation as formidable opponents was cemented in Roman accounts and popularized in stories like the opening of 'Gladiator.'",
Once we have interesting people—both historical and modern—the next step is making them fun to hold, look at, and play with. We use the OpenAI API to generate each person’s name, interests, and story based on their age, occupation, location, and era. Then a second API call produces an image that matches all those details. The result is a card that’s not just data—it’s a small, coherent character with a name, a backstory, and a visual identity. Kids and adults alike can pick it up, enjoy the graphics, and start imagining the worlds these people inhabit, past and present.
One thing we haven’t fully solved yet is representing income. For modern people, it’s relatively straightforward, but historical figures are trickier—there’s a lot of fuzzy math involved. We’ve tossed around ideas for future versions, like measuring earnings in ounces of gold per year instead of translating everything into modern dollars. It’s a small detail, but it highlights the tension between accurate data and playful representation. Beyond the people, the cards also teach about the world. Every card has a small map in the bottom right corner showing the location, and kids (and adults) can start recognizing countries, flags, and where different cultures existed. The cards also encode technology available to each person: not everyone has electricity or internet today, and in the past, access to longships, papyrus, or other tools varied widely. It’s a way to make geography, history, and technological development tangible while still keeping the game playful.
So far, we’ve mostly been talking about trading cards—people from different times and places, like a historical Pokémon. But what makes it a game? Keeping it simple enough for kids was key. I started sharing cards with teachers and administrators at my kid’s school, and we wanted something that’s easy to pick up and fun for adults, too. That led us to a new class of cards: event cards. Each event card has a number of victory points, and players win it by meeting requirements that are satisfied by the people cards in their hand. It’s open-ended and flexible, but it gives structure without overwhelming players.
On each event card, just below the image, location, and time, you’ll see a few colored rectangles with numbers labeled “civic,” “soldier,” “labor,” or similar. These are the requirements needed to win that card. Every person card—historical or modern—is stamped with one of these classes: engineers, finance, government, children, retirees, etc. Children and retirees introduce interesting gameplay dynamics. In the current model, a child can be raised into any class needed to meet an event card’s requirements, but it takes three turns. Retirees either get discarded or could accelerate raising a child, reducing it from three turns to one. It’s still open-ended, but these mechanics let us connect people to events in simple, strategic ways.
Supported Flags/Command Line Options
These scripts support the following flags:
program1_sample_positions.js
--places=<place1,place2,...>
Filter sampled positions to preferred places(by name,region,or description).
program2_sample_people.js
--historical-only
Only generate historical people(from eras inhistorical_eras.js).
--modern-only
Only generate modern people(no erafield).
A natural question is what happens if you sample the world “honestly.” Wouldn’t you just keep generating people from India and China today, or endless farmers in the past, or office workers on laptops if you zoom into the Bay Area? In practice, that’s exactly what naïve sampling gives you. The workaround is simple but powerful: command-line flags. Instead of sampling the entire planet, you can constrain the distribution—only sample New Zealand and Greece, tilt the mix toward historical or modern people, restrict to certain classes, or even specific eras. The system can still do pure random sampling, but these controls turn it into something more like a telescope or a microscope. You’re not choosing the person, just where and how closely to look.
So what’s it like to actually play the game right now, and did it meet our goals? The mechanics are still evolving. The core loop is simple, but one clear piece of feedback is that we should lean harder on the data already on the cards—things like economy, interests, and age, which right now only really matter for children. There’s a lot of room to build mechanics that make those fields meaningful. Even in its current form, though, the game is genuinely fun. We’ve played it with groups of friends, with and without kids, and people get into it quickly. Games move fast, and it doesn’t feel heavy or didactic.
As for the original motivation—helping kids form a clearer picture of the world—it’s not a silver bullet. This isn’t going to solve social cohesion or prevent us from sliding into some Mad Max future. But it does encourage a less aggressive, more cooperative way of thinking about people, across time and place. It shows that societies, cultures, and regions ebb and flow, and that history isn’t a snapshot—it’s a process. For us, as parents and builders, that felt like a worthwhile outcome.
Ever since my time in a crystallography lab, I’ve been fascinated by the process of translating raw diffraction patterns into detailed structural models.
I recently won an auction for a fairly worn, but very nostalgic 80s BYTE magazine. Don’t you just love the hand drawn artwork?
Back then, we relied on those cool SGI (Silicon Graphics, Inc.) computers—high-performance machines that were the go-to for graphics and visualization in the late ’90s and early 2000s. These workstations were the backbone of many scientific labs, enabling us to visualize complex molecular structures in ways that were groundbreaking at the time.
Even at that time, I was captivated by the pursuit of renders that were more than just scientific models—they were almost eye candy, visually arresting representations of the intricate dance of atoms. These weren’t just static arrangements; they were the keys to understanding quantum-scale processes. I found myself wondering: What do these molecular machines really look like? Are they transparent? Gooey?
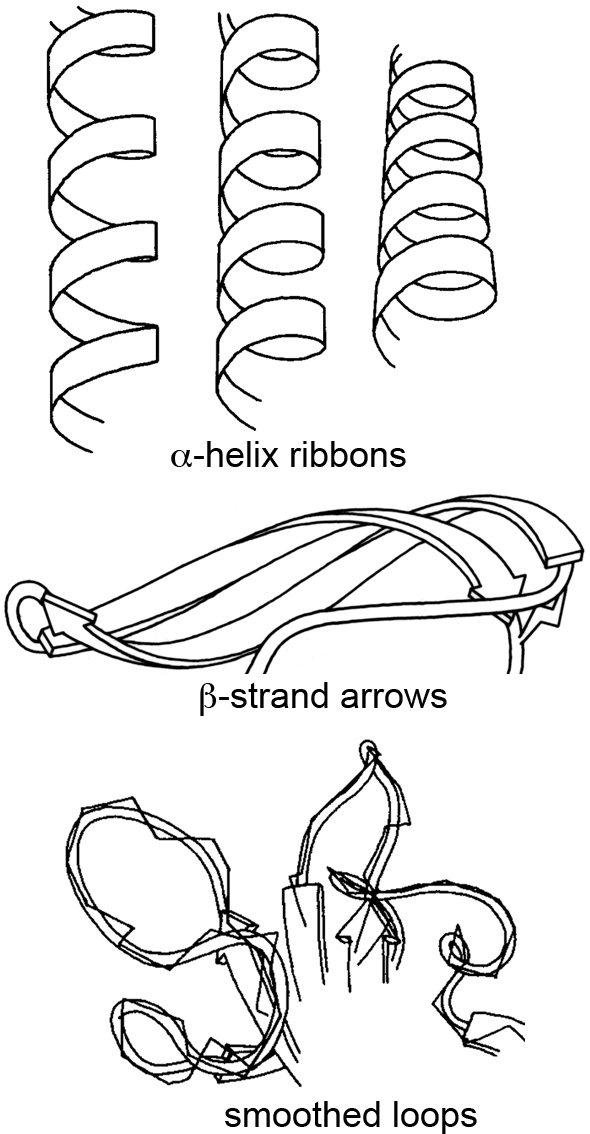
At the time, most people stuck with the ball-and-stick method of molecular rendering, and sometimes the very creative ribbon method for visualizing secondary structures like α-helices and β-sheets. This ribbon method, first introduced by Jane Richardson in the 1980s, revolutionized how we perceive and depict the elegant architecture of proteins.
I was particularly struck by a talk she gave a few years ago at the Machine Learning in Structural Biology (MLSB) workshop, which is part of the broader NeurIPS (Conference on Neural Information Processing Systems). The presentations at MLSB highlight how the intersection of machine learning and structural biology is opening up new avenues for visualizing and understanding complex biological data—building on the foundations researchers laid with creative solutions like the ribbon diagrams decades earlier. You can still view a recording of the entire Richardson talk here.
Fast forward a few years, and I find myself reminiscing about the times when Sonic Youth was the soundtrack to my late-night coding sessions, and I was drawn to ideas about being in the flow, letting go, and just being present in the moment. These days, hanging out with my wife and child brings that same sense of peace and connection. But as much as I cherish these moments, there’s something uniquely exhilarating about attending conferences, where you get to “talk shop” and be exposed to the latest innovations.
I remember the first time I saw molecular renderings by Drew Berry—it was at a VIZBI conference, a gathering that perfectly blends scientific rigor with creative visualization. VIZBI (Visualizing Biological Data) is more than just a conference; it’s an international meeting point for the best minds in science, bioinformatics, and data visualization. What makes VIZBI special is its emphasis on both the scientific and artistic aspects of data visualization.
The conference not only showcases cutting-edge visualizations that transform how life scientists view and interact with data, but it also encourages a deeper appreciation for the aesthetic quality of these visualizations. This was evident in Drew Berry’s work, which brought the molecular world to life in a way I had never seen before. The structures didn’t just sit there; they vibrated, darted around, and had this incredible “stochastic” feel to them, capturing the chaotic energy that defines molecular interactions. It was like seeing the molecules not just as static models, but as living entities, each with its own rhythm and motion. VIZBI isn’t just about keeping up with the latest research; it’s about being inspired, about seeing the boundaries of science and art blur in ways that open up new possibilities for how we understand life at its most fundamental level. It’s the kind of experience that reminds me why I got into this field in the first place.
In the last couple of years, BioVis has really stepped up its efforts to engage the community, working closely with other organizations like IEEE and ISCB to keep the spirit vibrant and interdisciplinary. BioVis has taken on the challenge of pushing forward the frontiers of biological data visualization, encouraging collaboration across fields and nurturing a community that is as diverse as it is innovative.
By bringing together visualization researchers with biologists and bioinformaticians, BioVis has managed to keep the conversation fresh and evolving, ensuring that new methods and ideas keep flowing. It’s exciting to see how these gatherings—both old and new—continue to energize the community and drive progress in understanding the complexities of life at every scale.
And hey, these are just some of the meetings out there, but they are my personal favorites. Even with incredible advancements like AlphaFold, RoseTTAFold, and ColabFold, which have made huge leaps in predicting molecular structures, there is still something uniquely thrilling about the art of representation. For me, that thrill is often fueled by the same sense of awe I get from playing video games. A good game isn’t just about throwing the newest engine at it; it’s about the aesthetics, the art, and the way it all comes together. That’s what makes good games age well—and I think it’s the same with science.
As the saying goes: sometimes, it’s less about where the path leads and more about the wonder found along the way.
Twenty years ago, my spouse-to-be and I met and became friends. During the pandemic, we got engaged and wanted to create a unique and memorable wedding invitation for our friends and family.
Due to a chip shortage, we used both Adafruit EdgeBadge and PyBadge LC to create interactive invitations. These credit card-sized boards are perfect for this purpose, boasting a range of impressive features. Each board includes a 1.8″ 160×128 color TFT display connected to its own SPI port, eight game/control buttons with nice silicone button tops, a triple-axis accelerometer for motion sensing, and a light sensor that points out the front. They also have a built-in buzzer mini-speaker, a mono Class-D speaker driver, a LiPoly battery port with built-in recharging capability, a USB port for battery charging, programming, and debugging, and two female header strips for potential expansions. Additionally, the EdgeBadge supports TensorFlow Lite for Microcontrollers, enabling machine learning features such as voice commands and gesture controls.
The game was developed using Microsoft MakeCode Arcade with JavaScript. Unique sprites were created using online sprite generators and tweaked for enhancement. Generic assets like trees were sourced from MakeCode Arcade. We also commissioned The Time Cowboy – Jake Lawrence, an Australian Cartoonist / Storyboard Artist, to create the box art and website art, some of which we turned into sprites for a cohesive design.
Designing a game for these low-cost, low-power devices presented its own set of challenges. Despite their capabilities, the first version of the game I created ran great on my MacBook, but the FPS was absolutely atrocious on the devboard! Initially, I thought about recreating the entire venue in the game to allow for an open-world experience, but it was too much for the board to handle.
I had to do some research and bust out some old video game tricks, like efficiently utilizing the same assets and dividing the map into many seamless “levels” that would be triggered and quickly loaded. This was both fun and challenging for someone who typically does bioinformatics work.
for (let index = 0; index < 8; index++) {
placedTre = sprites.create(
trz[tr_count],
SpriteKind.Food
)
placedTre.setPosition(
randint(40, 160),
randint(5, 30)
)
tr_count += 1
if (tr_count >= 4) {
tr_count = 0
}
}
Because I’m of North Indian Mughal descent, our wedding involved multiple days with different rituals, such as an entire messy day where we get covered in turmeric, and another day dedicated to having intricate patterns painted on us with henna. We thought a campground about 60 miles north of San Francisco would be perfect for these multiple days of celebration. This campground had many Airstreams, which we thought would be a comfortable way for our friends and family to spend the time, even if they weren’t familiar with camping.
The music for the game was also a fun project. For the end of the game, I created a MIDI cover of a Depeche Mode track:
“Come with me into the trees, We’ll lay on the grass and let the hours pass Take my hand, come back to the land Let’s get away, just for one day.”
All of this was actually inspired by DEFCON badges, and you can check out the finished game online at MakeCode Arcade, minus the hardware, in your browser. I highly recommend using these kinds of low-cost devboards as gifts to friends and family. They can create their own projects using them, which is also fun to see what people end up making.
Batches of invites ready to send.
Combining retro gaming charm with cutting-edge tech and cohesive design elements makes for an unforgettable and fun invitation. Try it out and make your invite the talk of the town!
Partner is pregnant, ultrasound looked cool. Had an e-ink dev board collecting dust. So here we are. Pretty simple stuff, the board is an Adafruit MagTag. And all we needed to do was have a progress bar, some graphics, and some text. There are some useful guides to making a progress bar and graphics for this e-ink board HERE and HERE.
I used some wikimedia fetal development graphics for each step. The key thing is to format the image correctly for the e-ink display. This is the ImageMagick command I used to do that:
Have it sticking on the fridge right now, the battery consumption is really low, and only used when updating the progress, should be good for a year or more.
Showing traditional carbon nanotube made only with carbon, versus microtubules found in cells made with Tubulin proteins.
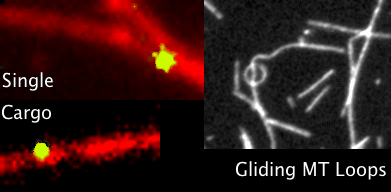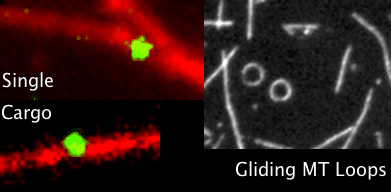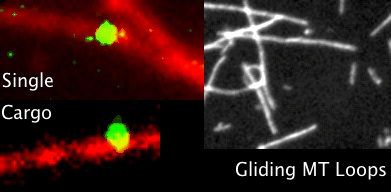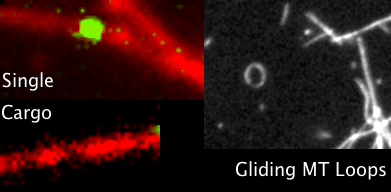
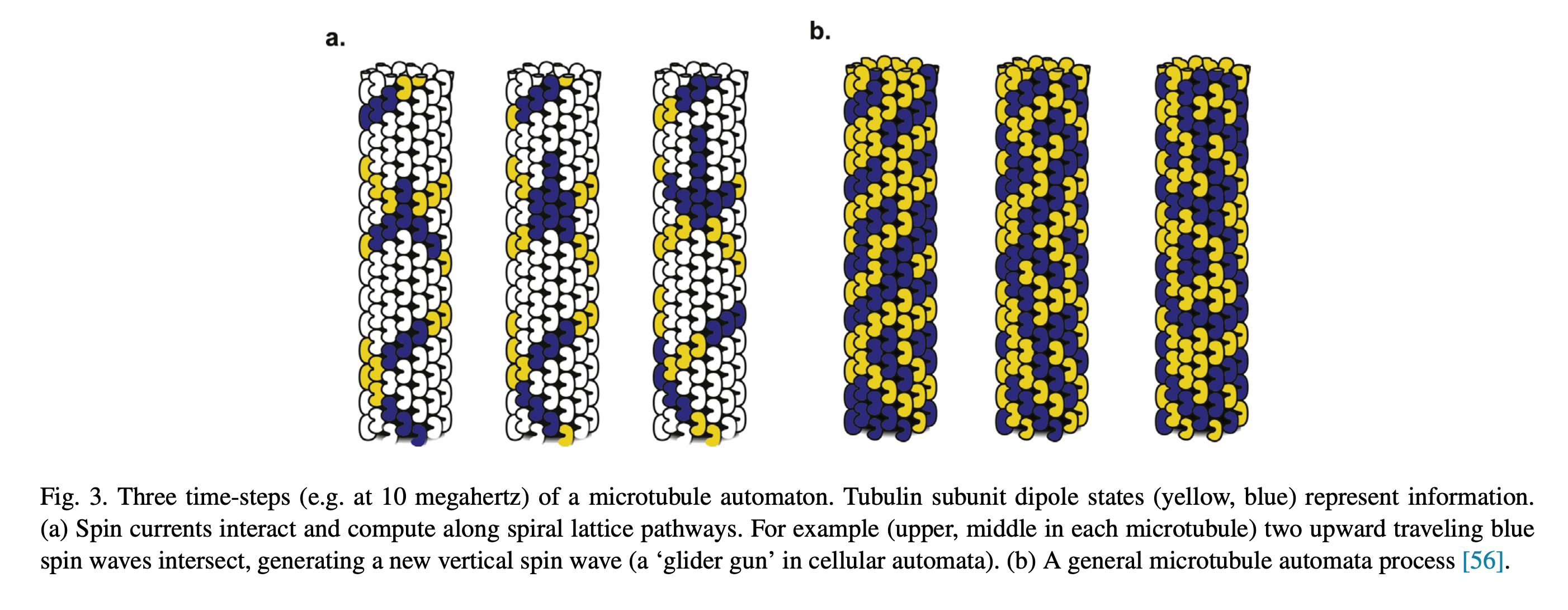
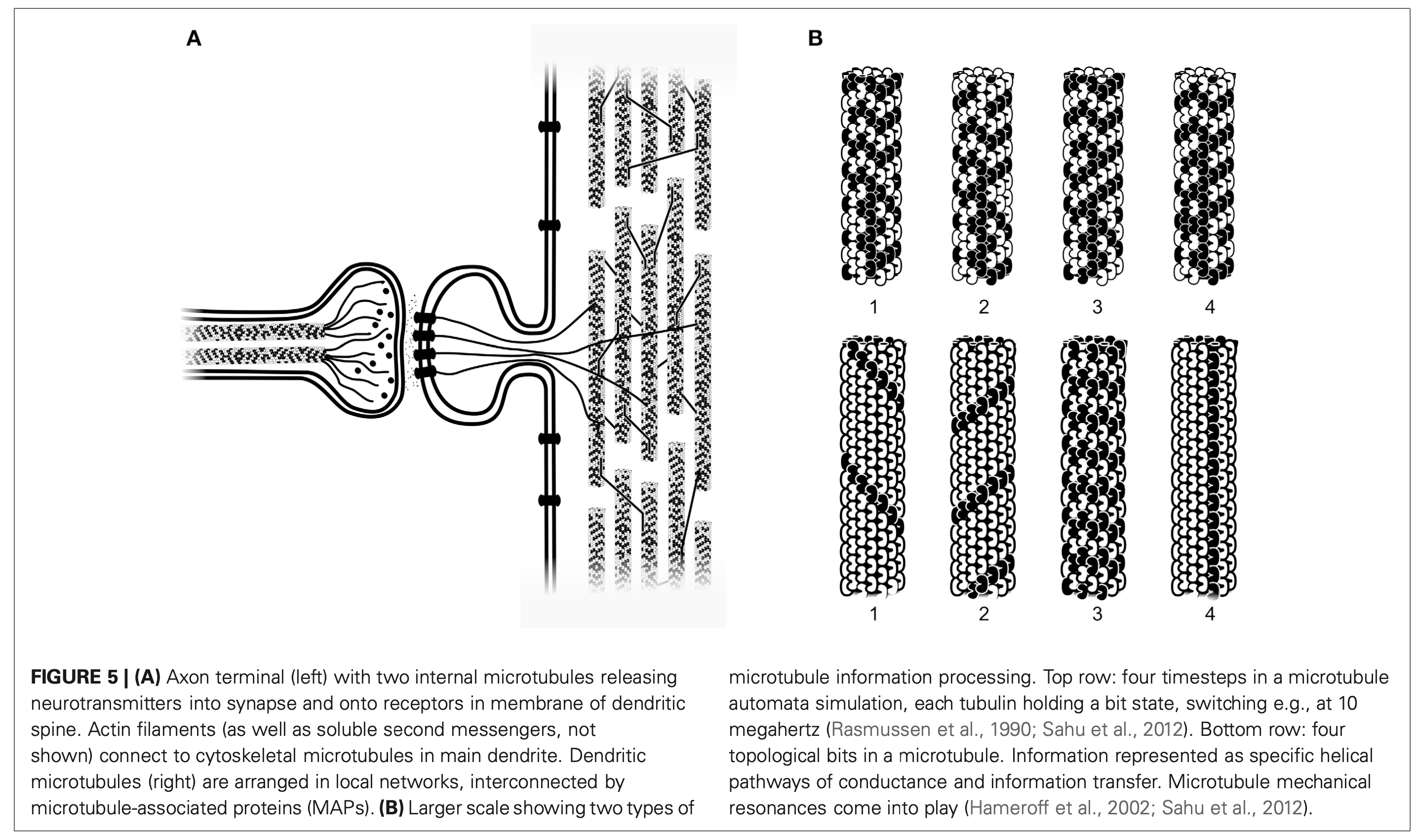
You’ve all heard of carbon nanotubes. You know, really small tubes made of just carbon atoms; maybe they’re good for moving electrons around. And supposedly carbon nanotubes might play a role in the next type of computers, so-called quantum computers. That’s carbon nanotubes. Then we have Microtubules, those with biology backgrounds will know about them, but for everyone else these are also really small tubes, but inside the cells of our bodies. We’ve seen microtubules do things inside cells even using microscopes, things like giving the cell types their shape; like a scaffolding. Microtubules have also been seen helping cells divide by grabbing things like chromosomes to opposite sides of a cell, and also as rails for transporting packages around cells. Anyway, the main point is microtubules do lots of things, we know this, but the question is do they also do some of the things that carbon nanotubes do?
Looking through the microscope to see some fun things microtubules do: pull chromosomes around, give cells their shape, act as rails for transport of goods.
Not only are microtubules hollow on the inside, say around 14nm which is similar to carbon nanotubes, they are also covered in a lattice of interesting molecules that act as binary switches. This is the G-protein signaling system, and it’s not exclusive to microtubules, rather it’s a family of molecules that work together within cells to create cascades of events and state changes. The outside of microtubules have GTP/GDP, Guanosine triphosphate and diphosphate. Removal of the 3rd phosphate is one state of the switch, while GTP is the other state. This lattice of GTP/GDP on the surface of the microtubules plays a known and observed role in vesicle transport, say when a “rail cart” attaches or detaches from the tubes, as well as in cell division, as in when the tubes attach to chromosomes; but what might be most fascinating are the fun patterns they form.
At this time for the hollow aspect of microtubules there has not been much, if any, observed behavior of electrons or other particles being transported within/through the tubes, only along the outside. But I may have just not seen those papers, or don’t know the right search terms. Some researchers have suggested, that these GTP/GDP lattices might themselves represent a form of computation, and/or electron transport. The most famous of these researchers is Roger Penrose, who is by no stretch a biologist, but there is some precedent of theoretical physicists getting close to the answer before evidence based molecular biology experiments validated the models. What comes to mind is the Trinity College lecture “What Is Life?” by Erwin Schrödinger, guessing characteristics of a molecule for transfer of hereditary information more than a decade before the structure of DNA was resolved.
Types of electron orbitals found in molecular bonds, with Pi-bonds at the bottom right, which is maybe what we are interested in.
Most bonds between atoms in molecules are either sigma bonds (single bonds) or pi-bonds (double bonds). And it seems like what Penrose and his friends are suggesting is related to a property of pi-bonds. We all know that at the center of atoms are protons & neutrons, they’re huge, and then electrons which are tiny orbit around that big center, like a planet and its moon, or a sun and its planets. But that’s really an oversimplified concept, and not really reality. As an atom gains electrons, from 1 to however many, each electron has a chance of being found in a certain region away from the center. So, yes the first 2 or 3 electrons might be found at any given time in a region that’s like concentric spheres around the atom but then things get weird. In double bonds (pi-bonds), which are common in organic molecules, a single electron can be found at the top half of the distribution or the bottom at any given instant; and when there are multiple pi-bonds in a row, like in many protein backbones, any of the electrons from any of atoms along the backbone can show up at any of the locations at any given instant. Or something, it’s weird, I don’t really get it, and maybe that’s the point Penrose is making. Quantum stuff is funky and doesn’t make much sense to dumb-dumbs like me.
There is a lot of excitement (hype) around Artificial Intelligence, all the great strides we have made with neural networks, and all the right people at the right venture retreats are saying general intelligence will change your lives forever. But much of the tech stack behind AI research is based on mimicking how neurons in biological systems are, and it may wholly be true that beyond synapses, dendrites, and neurotransmitters there is an entirely still hidden mechanism to intelligence. It may be what Penrose proposes, and it may not be. What’s clear is there is so much we do not understand about what happens within cells, on a molecular scale, and even less when it comes down to the subatomic scale. From what has been observed to date, living systems, even basic multicellular processes utilize these poorly understood mechanisms of particle behavior, let alone a process such as intelligence. Are microtubules the answer? Are carbon nanotubes and quantum processors? Is general intelligence right around the corner? Who knows. What seems important is to balance evidence based models with pure or more maths based models as guides. As is more common in physics, work it out on the chalkboard before/if ever diving in.
More and more, as we begin to get a solid grasp on DNA sequencing people are finding the need to understand what makes each type of cell different, or what changes occur before/after the introduction of a therapeutic. Of course, the answers are most likely in RNA, as the DNA is our permanent record, and the RNA is what is being worked with at any given moment. So, RNA sequencing has become more and more popular; however, trying to make sense of the data and actually understand what it is our machines are picking up has introduced a whole suite of challenges to overcome.
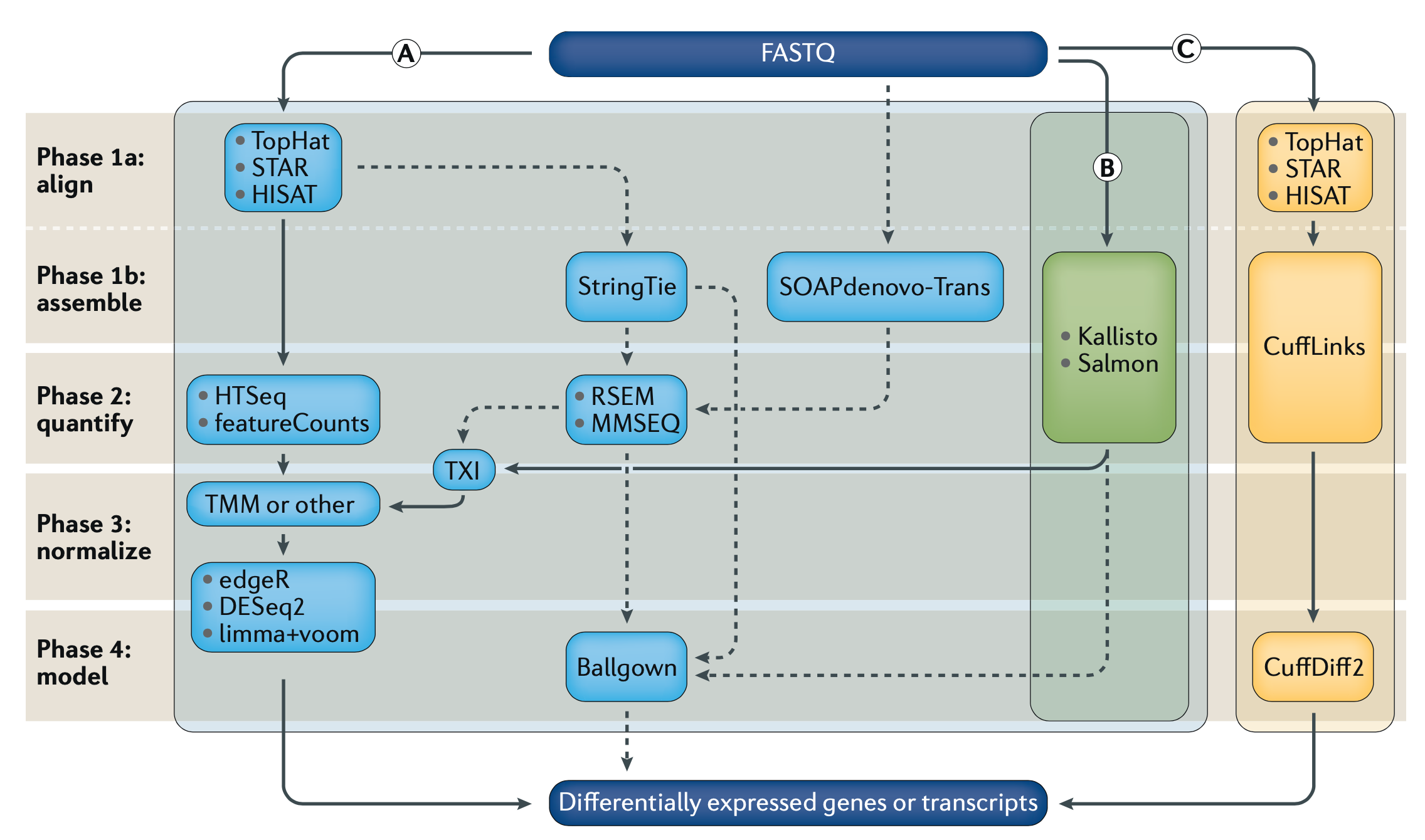
Differential Gene Expression (DGE) is currently the most common use for RNA-seq, where we try to find out which genes from our DNA are expressed differently across cell or sample types as RNA. Because the same machines by Illumina, PacBio, Oxford Nanopore, etc, are used to generate RNA sequencing data, and we need many more reads to get confident pictures of what’s happening across cells, DGE tends to be computationally expensive. As with DNA analysis, sequence alignment is the most time and resource consuming step. If we look at Fig1 above, we see three separate sets of algorithms, pipelines, to go from our raw data (FASTQ files) to our finished answers, we will focus on method (B).
Alignment-free analysis methods are a relatively new breakthrough, and allows us to take our sequencing data coming out of the machines, and skip over the worst part. There are new algorithms and tools coming out all the time, but Kallisto by Páll Melsted and Lior Pachter seems to be the winner for now. It’s also super easy to use. Depending on your OS one of the following commands will install it. Everything you see on this post was done on Linux AWS instances.
Ideally there’s some pre-processing that should be done on the FASTQ files with our RNA-seq data before jumping into the analysis but let’s leave that up to someone else to explain. Once Kallisto is installed, it will need a transcriptome index. This is very similar to the reference genome used in DNA analysis pipelines. The Pachter Lab provides several pre-built transcriptomes here including H.Sapiens. We can also build our own, after which there’s only one command to do the first part of our analysis.
Believe it or not, at this point Kallisto’s work is done. If we look in the output folder we can find the "abundance.tsv" file. This has our estimated counts of the number of our RNA-seq reads matched to their respective gene transcripts, essentially the more reads that are at a given gene the more that gene is being expressed in our sample/given cell. This is very basic, so there is a more statistically relevant number included in the file, Transcripts Per Million (TPM), which is something people love.
TPM simply shows the rate of counts per base (Xi/li) where we get a measurement of the proportion of transcripts in the pool of RNA, here it is in math.
There are other popular units of measurement like RPKM/FPKM, reads per kilobase per million reads mapped. These can be derived from TPM, so we can skip that for now and move on with our analysis, and the hard/not fun part for me, as shown in Fig1 after Kallisto we move to TXI, TMM, DESq2, etc. But instead we went with Sleuth, which is also made by the Pachter Lab.
Unfortunately, Sleuth is written in R and I hate R. Any way, I guess statisticians love it for their own reasons, so we’ll use it. Let’s start simple by installing Sleuth.
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install()
BiocManager::install("devtools") # only if devtools not yet installed
BiocManager::install("pachterlab/sleuth")
For the sake of building and testing this pipeline, we used 3 sample sets, where one is a tumor sample. Ideally, and the Hadfield et al paper talks about this, you want to use datasets of the same cell type, and have around 6 replicates. You know in an ideal world. Let’s just get the work done and get out of R as fast as we can.
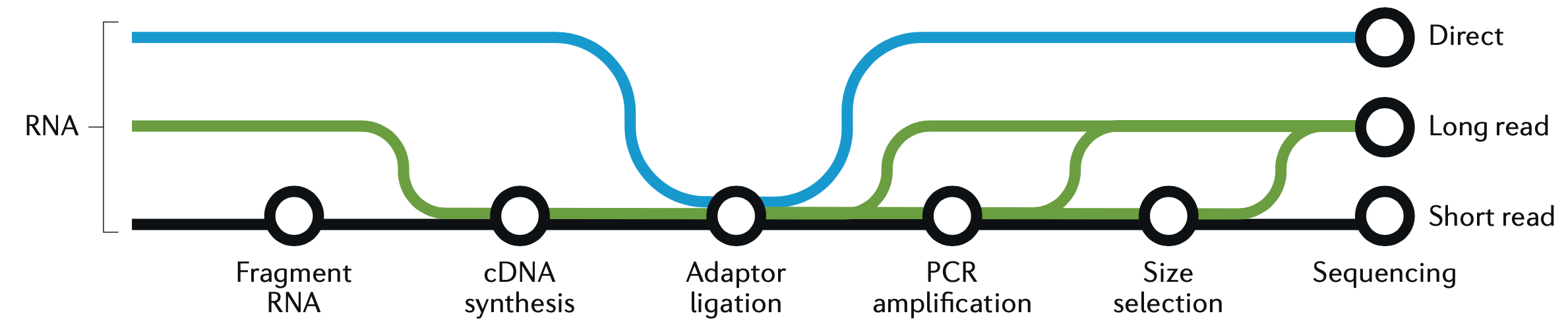
This is a good place to take a quick graphic break and look at another figure from the great Hadfield paper, before we continue with R and wrap up the analysis. This figure is a visualization of the different forms of RNA-seq reads, from long to short.
In the code block above we loaded Sleuth into R, named our samples, pointed R to our sample directories, added metadata, and actually ran our Sleuth analysis creating several models. Now let’s take a look at those models and filter the results before we make some plots.
We can see in the models, the data was analyzed based on cell type and whether or not there was any treatment. For the tests we can see that we have likelihood ratio tests as well as a wald test. Afterwards, we place all the results in a table, then we filter those and take our significant results. Mostly I like to filter based on a combination of the following parameters: var_obs: variance of observation, tech_var: technical variance of observation from the bootstraps (named 'sigma_q_sq' if rename_cols is FALSE), sigma_sq: raw estimator of the variance once the technical variance has been removed, smooth_sigma_sq: smooth regression fit for the shrinkage estimation. This seems to bring out differences, and separate out our data in a meaningful way, we can also look closely at the p-value and other parameters which can be found in the Sleuth manual. Using tail, we can see that in this particular case we went from a total of 188753 results, down to only 7. That’s a pretty good needle to hay ratio. So now we can plot our results.
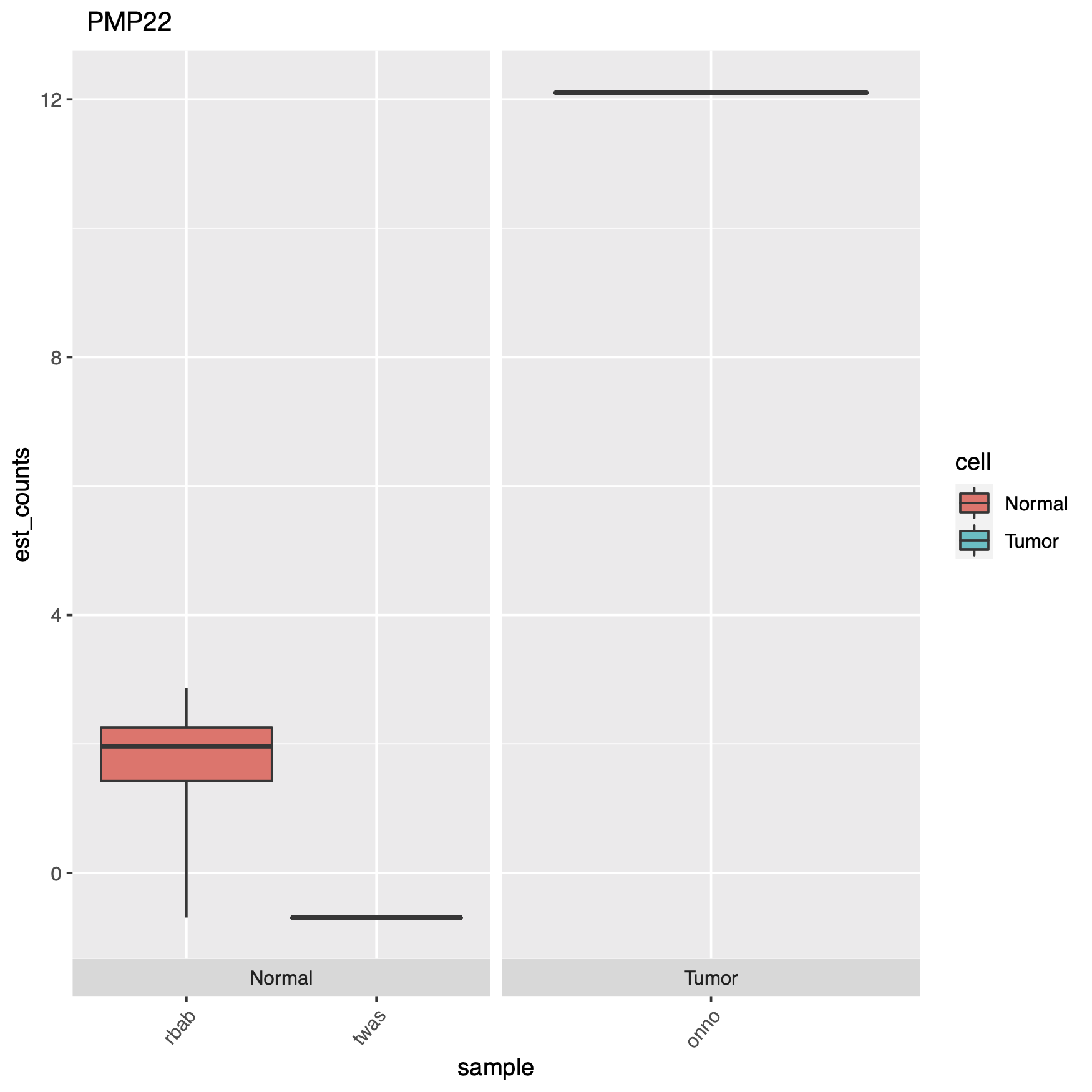
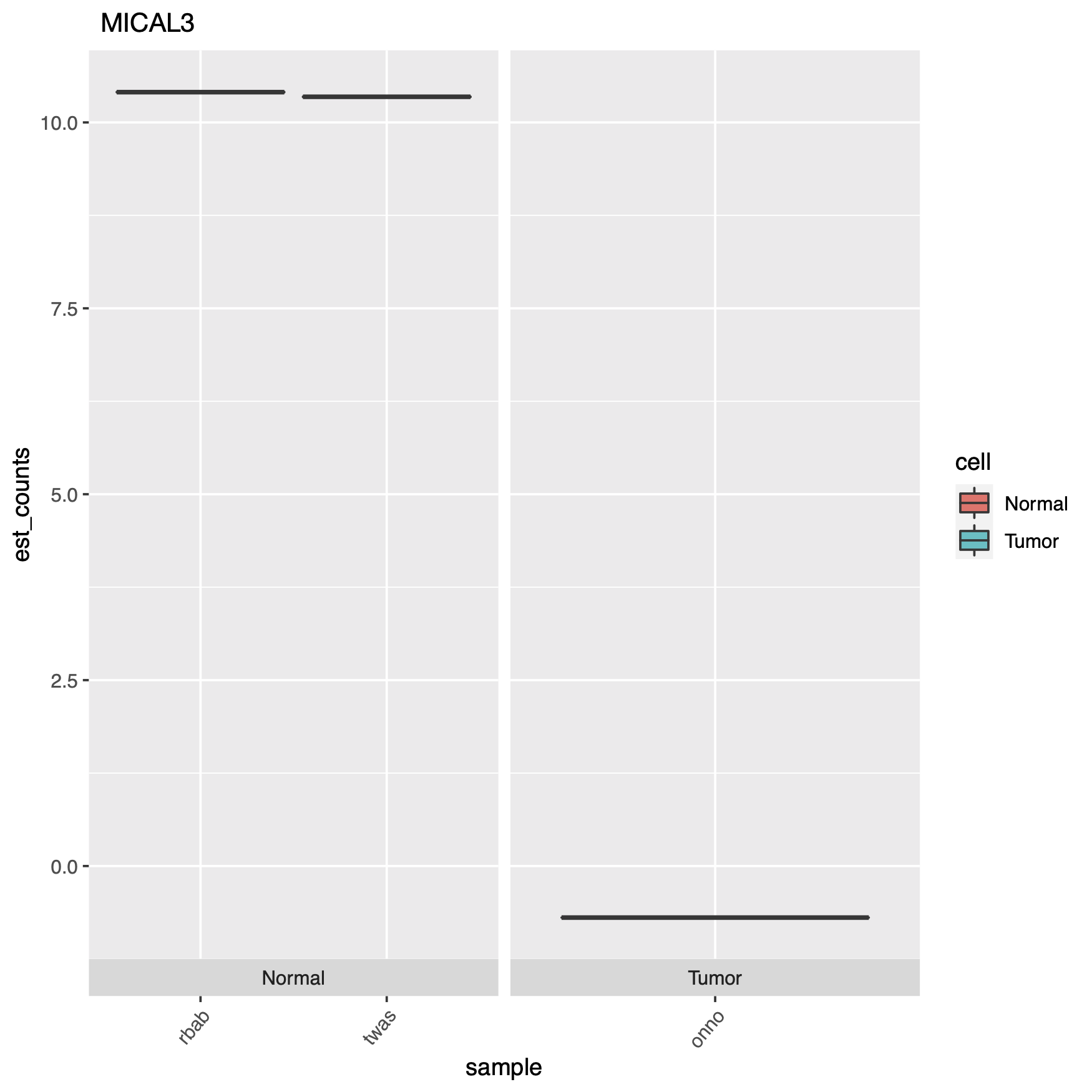
At the end of the code block above where we create our plots, there’s some extra code if you want to see actual gene names in your charts instead of the obscure transcript IDs, which can just as easily be converted with a google copy/paste. Here are some of the plots.
End of the day, this just demonstrates an easy way to set up an RNA-seq DGE analysis, and using a cool new technique which is alignment-free, this saves time & money. But remember it’s always important to have good experimental design, so generating the right data, meaning controls and replicates, as well as testing the right samples & cell types together. Look through the documentation the Pachter lab provides, read the Hadfield paper, and look around online for other sources of help, Harvard FAS Informatics has a good deal of RNA-seq guidance. Any way, good luck, R still sucks.
Can we use pipelines developed for human NGS analysis and quickly apply them for viral analysis? With ebolavirus being in the news, it seemed like a good time to try. Just as with a human sequencing project, it’s helpful if we have a good reference genome. The NCBI has four different ebola strain reference files located at their ftp:
Remote directory: /genomes/Viruses/*
Accession: NC_002549.1 : 18,959 bp linear cRNA
Accession: NC_014372.1 : 18,935 bp linear cRNA
Accession: NC_004161.1 : 18,891 bp linear cRNA
Accession: NC_014373.1 : 18,940 bp linear cRNA
Currently everything that’s happened in West Africa looks to match best with NC_002549.1, the Zaire strain. The Broad Institute began metagenomic sequencing from human serum this summer and the data can be accessed here (Accession: PRJNA257197). We can take some of these datasets and map them to NC_002549.1. The datasets are in .sra format, and must be extracted using fastq-dump.
Coverage map of SRA data from 2014 outbreak in Sierra Leone to the Zaire reference genome.
We can see that the data maps really well to this strain. All four of the reference genomes above were indexed with a new build of bwa(0.7.10-r876-dirty git clone https://github.com/lh3/bwa.git). Because EBOV genomes are so small, compared to humans, the only alignment algorithm which seemed suitable within bwa, was mem.
EBOV mokas$ ./bwa/bwa mem Zaire_ebolavirus_uid14703.fa SRR1553514.fastq > SRR1553514.sam
[M::main_mem] read 99010 sequences (10000010 bp)...
[M::mem_process_seqs] Processed 99010 reads in 8.988 CPU sec, 9.478 real sec
[M::main_mem] read 99010 sequences (10000010 bp)...
[M::mem_process_seqs] Processed 99010 reads in 8.964 CPU sec, 9.671 real sec
If we take the same SRA data and try to map it to some of the other strain references, e.g. the Reston Virginia strain from 1989, it can help give a rough idea of how closely related the 2014 incident is.
Very few regions from 2014 map to the Reston reference
It can be seen that apart from a few highly conserved regions where many reads align, the coverage map indicates that the data collected in West Africa and sequenced on the Illumina HiSeq2500 does not match to NC_004161.1. There were still approximately 500 variants with the Zaire reference on the 2014 samples, showing a good amount differences, considering the entire genome is only 18,000bp.
LucidAlign genome browser comparing the two strains
All of this is, of course, good news. We can take sequencing data of new EBOV strains and apply slightly modified pipelines to get meaningful results. And with the Ion PGM now being FDA approved means data can be generated in nearly 3 hours, with Federal approval.There have even been some publications which show that the protein VP24 can stop EBOV all together [DOI: 10.1086/520582] with the structures available for analysis as well. So, it looks like it’s all coming up humanity, our capabilities are there, and with proper resources this scary little bug can be a thing of history.
When it comes to genomics, contemporary bioinformatics follows the dogma of, assemble, detect, and annotate. This over-simplification however, washes over many key features, such as insertions and deletions, which may in fact be pathogenic[1].
Fig 1: A Low Complexity Region, featuring Homopolymers, leading to Ambiguous Mapping
High throughput, NGS short read data, and the algorithms developed to support them have overwhelmingly focused on detection of SNPs, while processing Structural Variants as an afterthought. This has largely been due to the combined limitations of chemistry and computation. Popular sequencers (I’m looking at you Illumina) produce strings with character lengths of several hundred bases. Matching millions of string-sets across highly-repetitive regions, where a single character or pattern of characters repeats over and over, is a nightmare from a algorithm design perspective.
Fig 2: Using Haplotype Calculations with Dindel, SeqAn, and Boost C++ Libraries to Resolve INDELs
Because scientists and engineers are a lazy bunch with deep-rooted personality flaws, we’ve brushed the problem of sequence alignment in repetitive and low complexity regions under the rug. Sure, most aligners and callers will have some implementation for INDEL realignment, and detection (say Picard or Haplotype Caller in GATK), but if you’ve ever seen the results you know they don’t hold up well to scrutiny. Amongst this, the work of Kees Albers stands out, and anyone who has written a validated pipeline will attest to the results by Dindel’s four step process[2]:
The process outlined above is quite compute intensive, and as can be seen in Figure 2, the number of possible haplotypes to resolve a single potential INDEL can range in the triple digits; each of which has to be aligned, and selected for by statistical relevance. Guillermo del Angel gives a great outline for those who use GATK (we find the results from Samtools more agreeable) to utilize Dindel, however even this has a fairly significant false positive rate[3].
Fig 3: GATK with Dindel Still has High FPR
An algorithm, regardless of how well designed, can’t overcome bad input data. And short reads are simply not good starting material for resolving structural variants. Luckily, several companies have taken on the challenge of developing long-read technologies, where the string lengths can reach tens of thousands of characters. We’ve been lucky enough to work with Pacific Biosciences, using data generated with their SMRT hardware and P5-C3 chemistry. Reads in suspected SV regions were extracted, the long reads were used to span the low-complexity/highly repetitive regions, and short reads reassembled using bayesian and HMM methods available in SeqAn and Boost.
At this point I should mention that while Boost is absolutely fabulous in-terms of providing great tools for C++ development, which is a great way to handle compute intensive tasks, it is without a doubt: pull-your-hair-out-frustraiting to build and use. Moreover, while our methods have significantly decreased the number of total SVs in our results, we haven’t had the chance to do a rigorous comparison for FPR; but I think a good way to do that would be similar to what Heng Li has shown recently[4].
Continuing from the previous post[1], dealing with structural effects of variants, we can now abstract one more level up and investigate our sequencing results from a relational pathway model.
Global Metabolic Pathway Map of H.sapien by Kanehisa Laboratories
The Kyoto Encyclopedia of Genes and Genomes (KEGG) has become an indispensable resource which has laboriously, and often manually, curated high-level functions of biological systems. Bioconductor, though not as essential as KEGG, provides some valuable tools when utilizing graph-theory for genomic analysis. If your data is well annotated and you happen to care about high-level genomic interactions, then you may have pathway annotations, containing data like the following: KEGG=hsa00071:FattyAcidMetabolism;
KEGG=hsa00280:Valine,LeucineAndIsoleucineDegradation;
KEGG=hsa00410:betaAlanineMetabolism;
KEGG IDs can be stored on an external file separate from the sequence data they are derived from. Though, storing the IDs with their respective variant is helpful, and it is possible to maintain VCF 4.1 specifications.
KEGG Annotations in VCF 4.1
As most Bioconductor tools are based on the R programming language, having an updated installation is recommended, this post uses version 3.0.1 “Good Sport”. Creating interaction maps with KEGG data will require three packages: KEGGgraph, Rgraphviz, and org.Hs.eg.db. These packages can be downloaded as separate tarballs, however installation from within R is likely best: $R
source("http://bioconductor.org/biocLite.R")
biocLite("KEGGgraph")
library(KEGGgraph)
Using the method above for all three. KEGG relational information is stored within XML files in the KEGG Markup Language. KGML files can be accessed through several methods, including directly from R, FTP, and subjectively the best method with REST-style KEGG API.
Bioconductor packages downloaded above come with a few KGML files pre-loaded, which can be viewed with the following command, it is also important to note that KGML files we want to use should be placed in this directory to avoid any unnecessary errors. $R
dir(system.file("extdata",package="KEGGgraph"))
$../Resources/library/KEGGgraph/extdata/
In this post the branched-chain amino acid (BCAA) degradation pathway, which has a KEGG ID of hsa00280, will be mapped in relation to variants from the BCKDHA gene. $R
[var1] <- system.file(".xml",package="KEGGgraph")
[var2] <- parseKGML2Graph([var1], genesOnly=TRUE)
[var3] <- c("[KEGG-Gene-ID]",edges([var2])$'[KEGG-Gene-ID]')
[var4] <- subKEGGgraph([var3],[variable2])
[var5] <- sapply(edges([var4]), length) > 0
[var6] <- sapply(inEdges([var4]), lendth) > 0
[var7] <- [var5]|[var6]
[var8] <- translateKEGGID2GeneID(names([var7]))
[var9] <- sapply(mget([var8],org.Hs.egSYMBOL),"[[",1))
[var10] <- [var4]
nodes([var10]) <- [var9]
[var11] <- list();
[var11]$fillcolor <- makeAttr([var4],"[color]")
plot([var10], nodeAttrs=[var11])
Executing these steps will result in a graph whose nodes and edges should help clarify any relevant connections between the genomic regions in question.
Results
While dynamic visualization tools (e.g. Gephi, Ayasdi, Cytoscape) look similar, and with some work utilize KEGG, they may lack the specificity and control which Bioconductor provides due to its foundations in R. These methods are necessary to understand more than just metabolic diseases, they also play a crucial role in drug interactions, compound heterozygosity/complex non-mendellian traits, and other high-level biological functions.







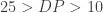 &
&  . However these expressions have to be written in Java Expression Language (
. However these expressions have to be written in Java Expression Language (


































 Can we use pipelines developed for human NGS analysis and quickly apply them for viral analysis? With ebolavirus being in the news, it seemed like a good time to try. Just as with a human sequencing project, it’s helpful if we have a good reference genome. The NCBI has four different ebola strain reference files located at their ftp:
Can we use pipelines developed for human NGS analysis and quickly apply them for viral analysis? With ebolavirus being in the news, it seemed like a good time to try. Just as with a human sequencing project, it’s helpful if we have a good reference genome. The NCBI has four different ebola strain reference files located at their ftp: