
Over christmas the Genome Reference Consortium gave all of us doing in silico life-science a wonderful present, or maybe it was just a lump of coal. GRCh38, the newest human reference genome assembly, was released to cheers and jeers abound.
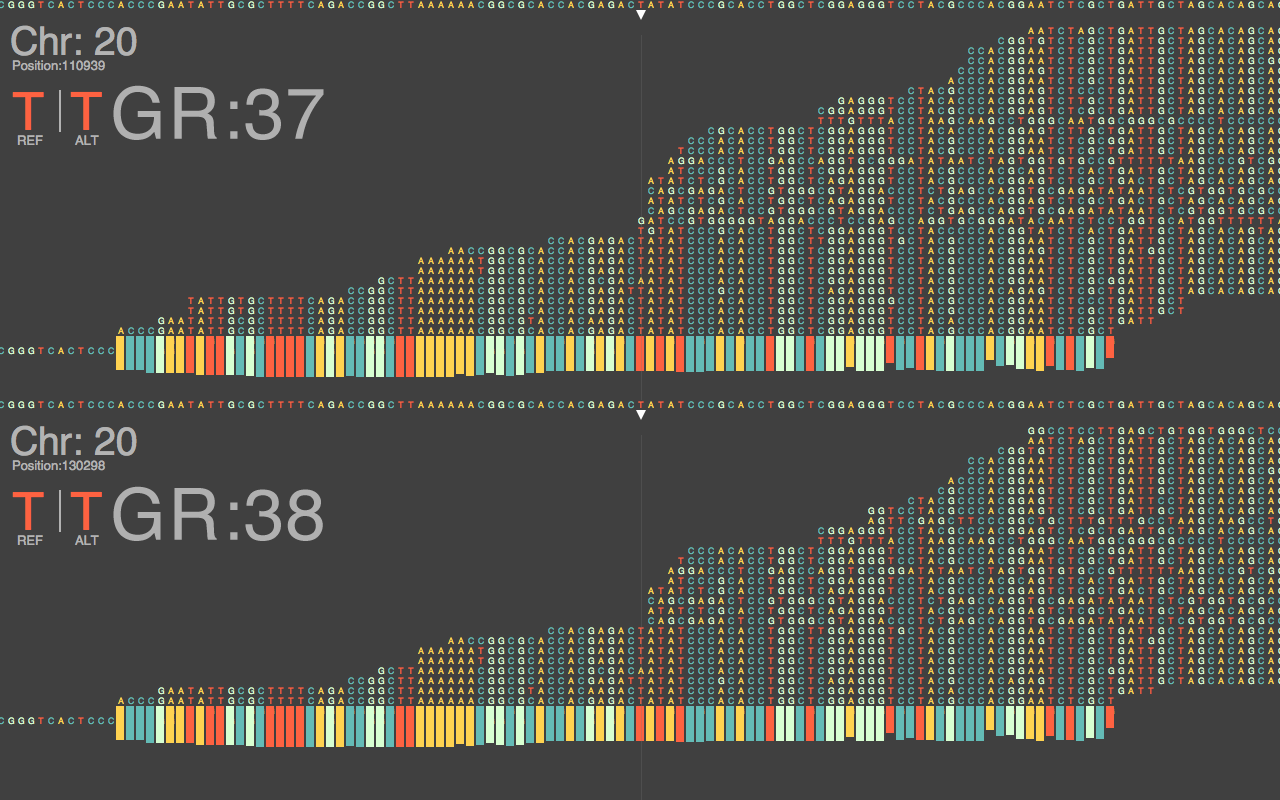
Fig.1: Chromosome 20 Assembly With BWA
Of course, most of us are excited to have up-to-date standards, especially with something like the human reference playing such a pivotal role in clinical adoption of genomics. However, some are lamenting the perhaps inevitable remapping of their NGS reads to this new reference. And it’s completely reasonable to have this worry. Will previous results from samples remain valid once assembled with this new reference, and how different will the sequence alignments be?

Fig.2: Ts/Tv Ratios between GRCh37 & 38
It will take months and years to thoroughly answer these questions, and notice the complete impact of this update on current NGS data. Consequentially, this does not keep us from starting to get preliminary ideas of what to expect in the coming years of working with this build. Figure 1 above, shows a nucelotide-level closeup of a BWA sequence alignment of the same dataset, generated on a HiSeq 2500, across the last major release of GRCh37 and the new GRCh38.
Internally we have been using chromosome 20 of human reference builds to benchmark tools and pipelines with datasets; it has a favorable size in terms of length, and not too many curveballs in terms of features. Figure 2 to the left, shows the Ts/Tv ratios between the two alignments of the same data across the two references to be quite similar at 0.3527 and 0.3445, respectively. Working with the slew of aligners, BWA has repeatedly shown itself to produce reliable results, while avoiding any overly-complex algorithms and trendy implementations. It’s a good workhorse.
Similarly, samtools was used to parse through our SAM/BAM files to produce VCFs with mpileup. Which, again, does not have the most bells and whistles, but is consistently reliable and good for comparing a single variable, in this case, the reference.

Fig.3: High-level Alignment Map Topology
Quantifiably, GRCh38 is very similar to the later GRCh37 releases, showing a change rate of 1 change every 159,558 bases on 37.69 and 1 change every 156,779 bases on 38 for our chromosome 20 dataset. Which, to use a technical term, looks pretty damn close. One of the major updates according to the GRC are changes to chromosome coordinates, which some back of the envelope math seems to give a Δ of +19,359 between GRCh37.69 and 38. In combination with one of the other major updates, sequence representation for centromeres, short-reads appear to be spread thinner to cover this difference, resulting in slightly lower depth of coverage versus 37.69. Figure 3 above, shows that overall the alignment map remains mostly similar, at least when BWA is used with standard Illumina reads; with somewhat negligible loss of DP.

Fig.4: Chr20 Annotation of Regional Features
By far the largest noticeable change brought to preexisting datasets appears to be related to annotation. Figure 4 above, shows just how hopelessly incongruent GRCh38 is at the moment with current annotation resources, yielding the largest differences to the latest GRCh37 assembly for the same reads. But this was to be expected, annotation will very likely be the last to catch up to this new build, and will improve over months and years.

So, should your project start using GRCh38? The short answer is, not yet. The long answer, it depends on your project resources, pipeline flexibility, and the questions trying to be answered. It’s helpful to remap preexisting NGS data to this new reference, and newly generated datasets would benefit the most, as sequence alignment tends to be the most expensive process in the pipeline to redo. Just keep in mind that for useful results your entire analysis pipeline, which is often an amalgamation of various opensource, commercial, and internal components has to work together. For the time being, GRCh38 is a wrench in the gears for many people, but it has a very promising future.






 &
&  . However these expressions have to be written in Java Expression Language (
. However these expressions have to be written in Java Expression Language (






















")




