
While the UnifiedGenotyper included within the Genome Analysis Toolkit (GATK) provides an ample method by which to call SNPs and indels, mpileup within Samtools still remains a reliable, quick and straightforward way to get variants.
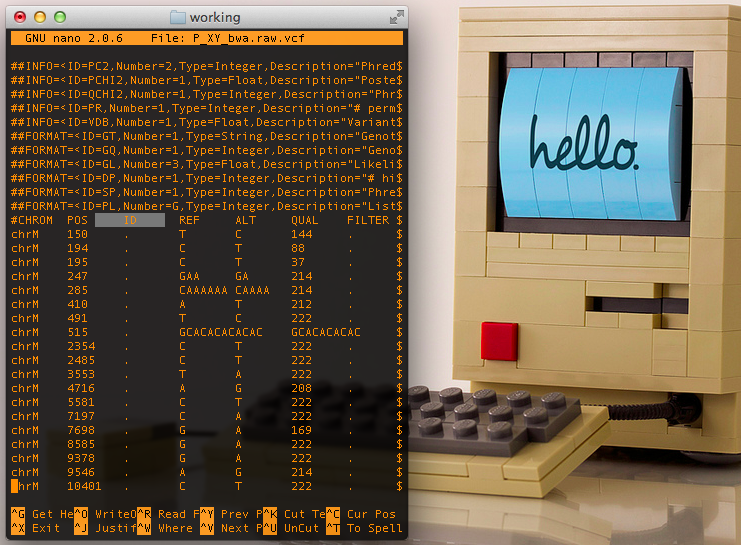
Raw VCF file from Samtools, notice lack of annotations & filters in the 3rd and 7th columns
To begin we take our assembled bam files created by the method of your choice, two of which are described in the previous posts[1][2]. With newer versions of Samtools the pileup function is replaced by mpileup, they perform the exact same actions; however, in traditional pileup we pass a single individual genome as a bam file for variant discovery, while in mipleup we can pass multiple individuals together and each of their variants are discovered within a single file as the output.
$samtools mpileup -uf [reference.fa] [.bam 1] [.bam 2] [.bam...] | bcftools view -bvcg -> [raw.variant.bcf]
$bcftools view [raw.variant.bcf] > [raw.variant.vcf]
Even though we’re saying the variant discovery is by samtools, all the actual work is being done by bcftools. To learn more about what bcftools can do check out the documentation, all the modules are included as a subdirectory within the samtools package.

Now that we have a VCF file containing all the positions where our samples differ from the reference, and each other, we can begin to utilize the appropriate GATK modules. Starting with annotation:
$java -Xmx[allocate memory] -jar GenomeAnalysisTK.jar -T VariantAnnotator -R [reference.fa] --variant [raw.vcf] --dbsnp [db.vcf] -L [raw.vcf] --alwaysAppendDbsnpId -o [annotated.vcf]
As you can see these one liners can get quite long, but rest assured, the results are worthwhile. If you look carefully at the above command you can see that we’re annotating based on a second VCF file, which in this case is being attained from the NCBI’s dbSNP. Feel free to use whatever database you see fit to generate your annotations.

Annotated VCF, notice rsIDs in 3rd column
Annotating our raw VCF with a dbSNP file results in flagging any polymorphisms between our sets to be marked with an rsID. These unique identifiers are used to track individual disease phenotypes, which are at various points of experimental validation. However, if we take our mapped genome and search for variations we’ll soon find that there are simply too many variations that show up to make any sense of our data. We have to decrease the size of our haystack before we start looking for our needle. This is where Filtration comes in. A high-level overview of the process can be seen in this previous post which utilizes a key figure from Genomics & Computation (available on iTunes). Below we execute a part of these concepts using GATK:
$java -Xmx[allocate memory] -jar GenomeAnalysisTK.jar -T VariantFiltration -R [reference.fa] --input [input.vcf] -o [output.vcf] --filterExpression "[insert expression]" --filterName "[expression name]"
It is important to understand the one-to-one mapping of filtering expression to the filter name to adequately use this module. A filterExpression should take any number of fields available within the INFO field for any given variation, such as:
AC1=1;AF1=0.5005;DP=130;DP4=3,0,4,3;FQ=3.02;MQ=44;PV4=0.47,0.038,1,0.19;VDB=0.0253
For example the expression could take into account the depth of read as well as the mapping quality, stating 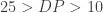


Trio Variant Visualization w/ HivePlot
There are many more steps towards refinement, i.e. recalibration and variant selection, but this blog post is getting quite long. And I think if you follow the roadsigns laid out here the full abilities of both Samtools & the GATK will become evident. The final payoff being reliable, meaningful, and thus useful, NGS data. Hit me up if you get stuck or think my ways are lame.



